

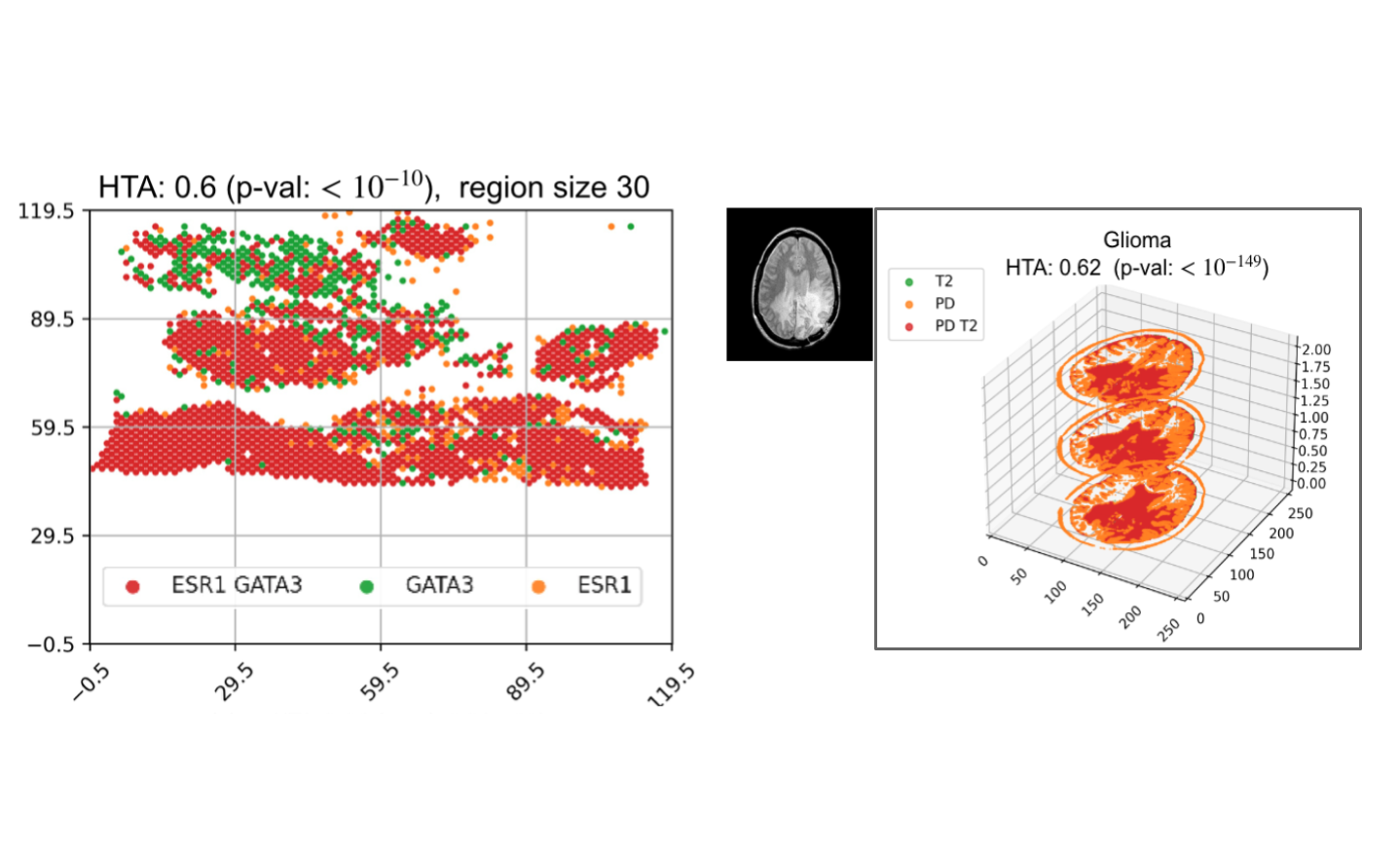
Assessing heterogeneity in spatial transcriptomics and imaging data using the HTA index
Emerging spatial transcriptomics data hold the potential to further our understanding of tumour heterogeneity and its clinical implications. Existing statistical tools are not sufficiently powerful to capture heterogeneity in the complex setting of spatial molecular biology. In this work, we developed the HeTerogeneity Average index (HTA), specifically designed to handle the multivariate nature of spatial transcriptomics. We prove that HTA has an approximately normal distribution, lending itself to efficient statistical assessment. We demonstrate how HTA can be used to analyse heterogeneity in two cancer spatial transcriptomics datasets: spatial RNA sequencing by 10x Genomics and spatial transcriptomics inferred from H&E. We also demonstrate that HTA applies to 3D spatial data using brain MRI scans.
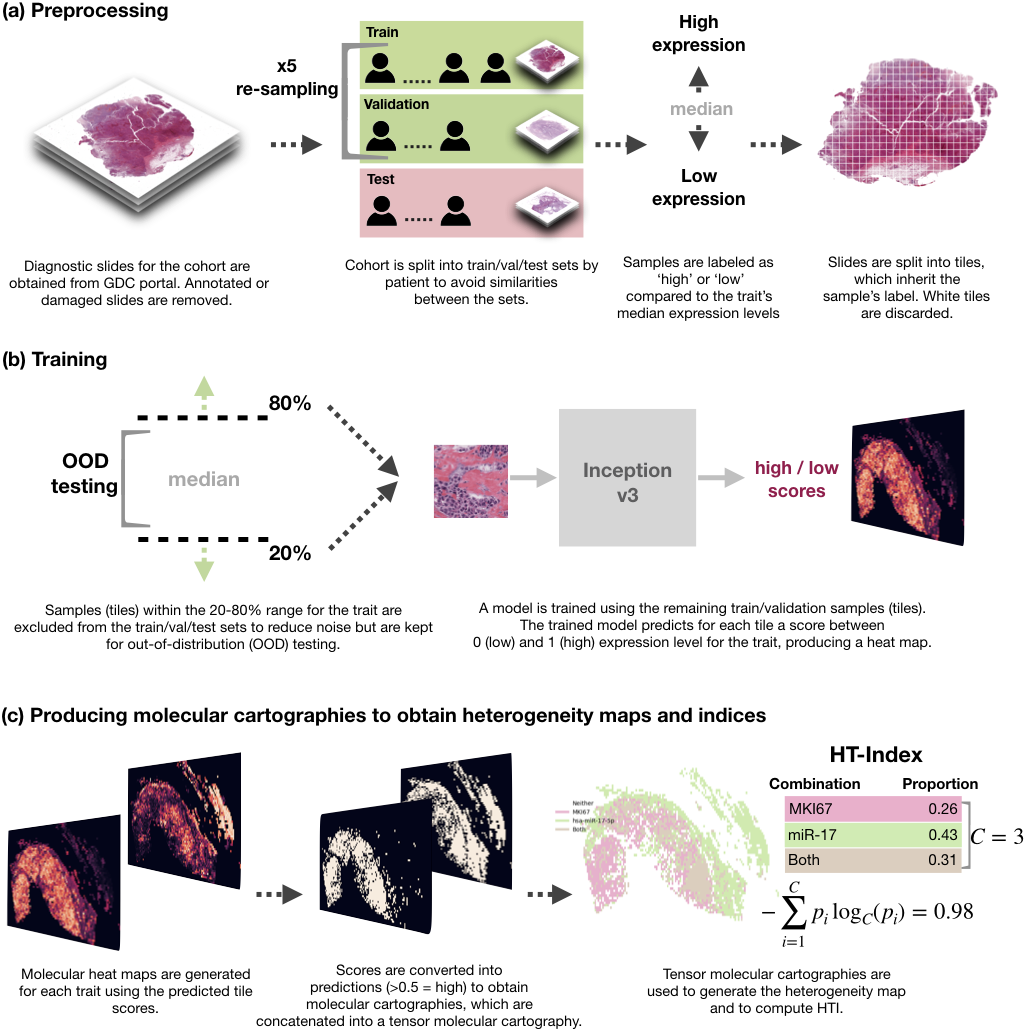
Spatial transcriptomics from pathology whole-slide images
In this work, we trained deep learning models to spatially resolve bulk mRNA and miRNA expression levels on pathology whole-slide images. Our models reach up to 0.95 AUC on held-out test sets from two cancer cohorts using a simple training pipeline and a small number of training samples. We also developed a method to spatially characterize tumor heterogeneity from the predicted gene expression levels. Applying our methods to breast and lung cancer slides, we discovered a significant statistical link between heterogeneity and survival. This project was supported by the European Union's Horizon 2020 and Google Cloud funding.
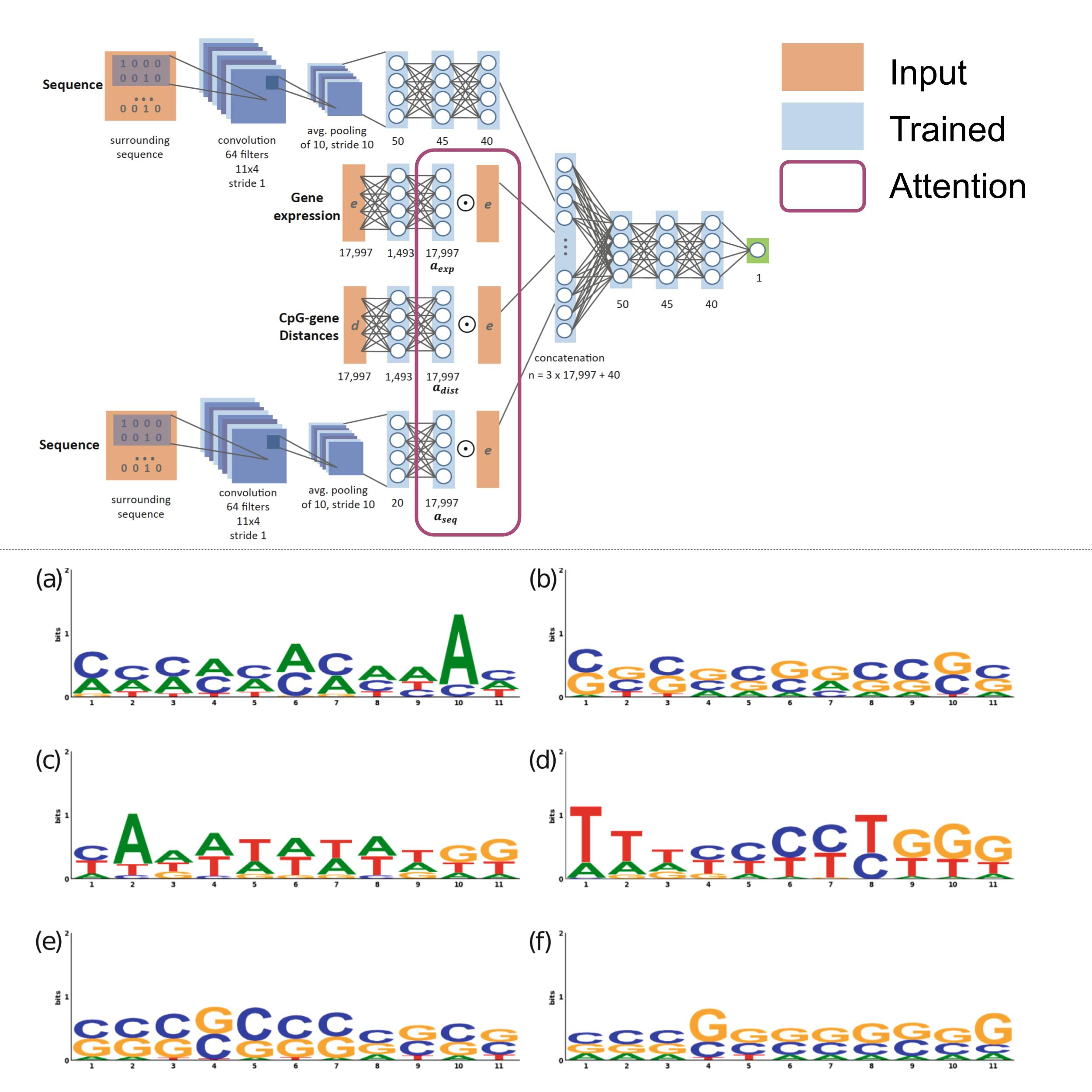
Predicting DNA methylation using deep learning with attention
DNA methylation has been extensively linked to alterations in gene expression, playing a key role in multiple diseases, especially cancer. In this work, we designed a deep learning network with an attention mechanism to predict methylation solely from sequence and gene expression. We also developed a framework to analyze the learned representations, which led to the discovery of novel links between methylation and gene expression. Our regression model obtains a Spearman correlation of 0.84 for thousands of CpG sites on two separate test sets and links several motifs and genes to methylation activity. The figure shows several DNA motifs identified by the network, including the TATA box and CpG islands, both known to play key roles in determining gene expression levels.
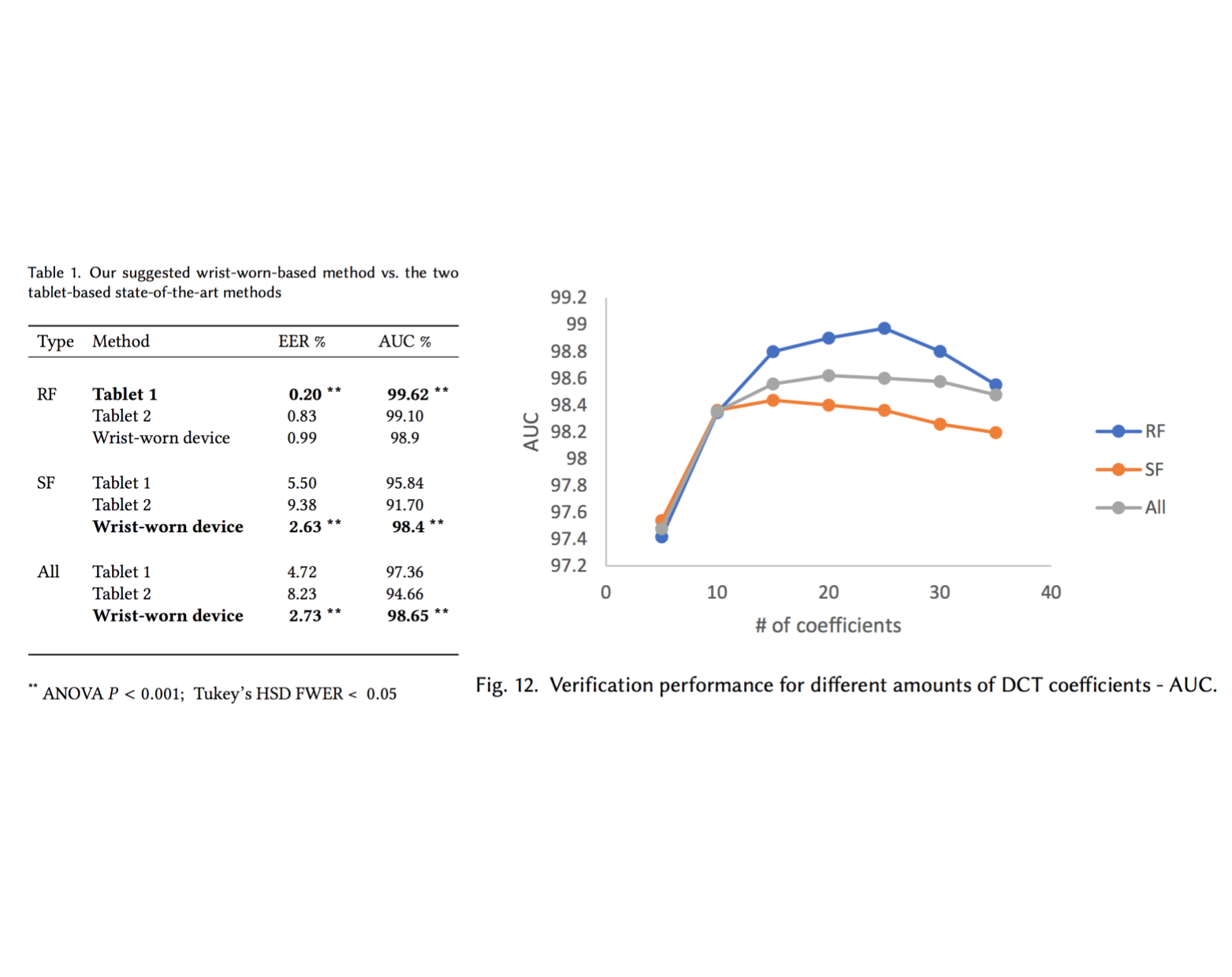
Handwritten signature verification using wrist-worn devices
In this work, we developed a machine learning model that identifies forged signatures based solely on motion signals captured from a smart-watch device (accelerometer, gyroscope etc.). To collect the data, we developed an android application that connects to a Microsoft Band 1, and used it to capture hundreds of signatures from 60 subjects. Our model significantly outperforms two state-of-the-art systems in skilled forgeries (SF), obtaining an EER of 2.63% and an AUC of 0.984. This paper won an outstanding student paper award (3rd place) in The 20th Israeli National Conference on Industrial Engineering.